Kügler lab
Research in our laboratory is based on two major pillars:
- understanding the pathophysiological mechanisms causing neurodegenerative disorders like Parkinson´s disease. Special attention is paid to proteins of the synuclein family, since a-synuclein is a major component of aggregate structures that form a hallmark of this disease. Blood-borne immunoglobulins against synucleins may invade the brain through an impaired blood-brain-barrier, thereby linking heart and brain science.
- development of adeno-associated viral vector (AAV) technology, enabling us to express any kind of protein or regulatory RNA in cells refractory to conventional gene transfer methods both in vitro and in vivo. AAV vectors are used for basic science as well as for establishment of pre-clinical gene therapy protocols. While we focus on CNS disorders, collaborative projects address diseases of the heart and the muscular system.
Vector tech and general applications
We develop and use AAV viral vectors for a wide variety of applications, i.e. for expression of Parkinson´s disease related proteins and genetically encoded sensors (e.g. for quantification of levels of calcium, ATP, ROS, cAMP etc.), both in cultured neurons andin the rodent brain. Expression of proteins or regulatory RNAs can be targeted to specific cell types or to specific sub-cellular organelles. The AAV toolkit offers extraordinary efficacy, safety and flexibility, in basic science but also in development of gene therapy.
For many internal and external collaborations, we have manufactured a large number and variety of AAV vectors (some of which reported here: ADDIN EN.CITE.DATA ADDIN EN.CITE.DATA (1-36)), while lentiviral (LV) vectors are rarely used (37) and adenoviral (AD) and Semliki-forest-virus based vectors (SFV) have meanwhile been fully discontinued.
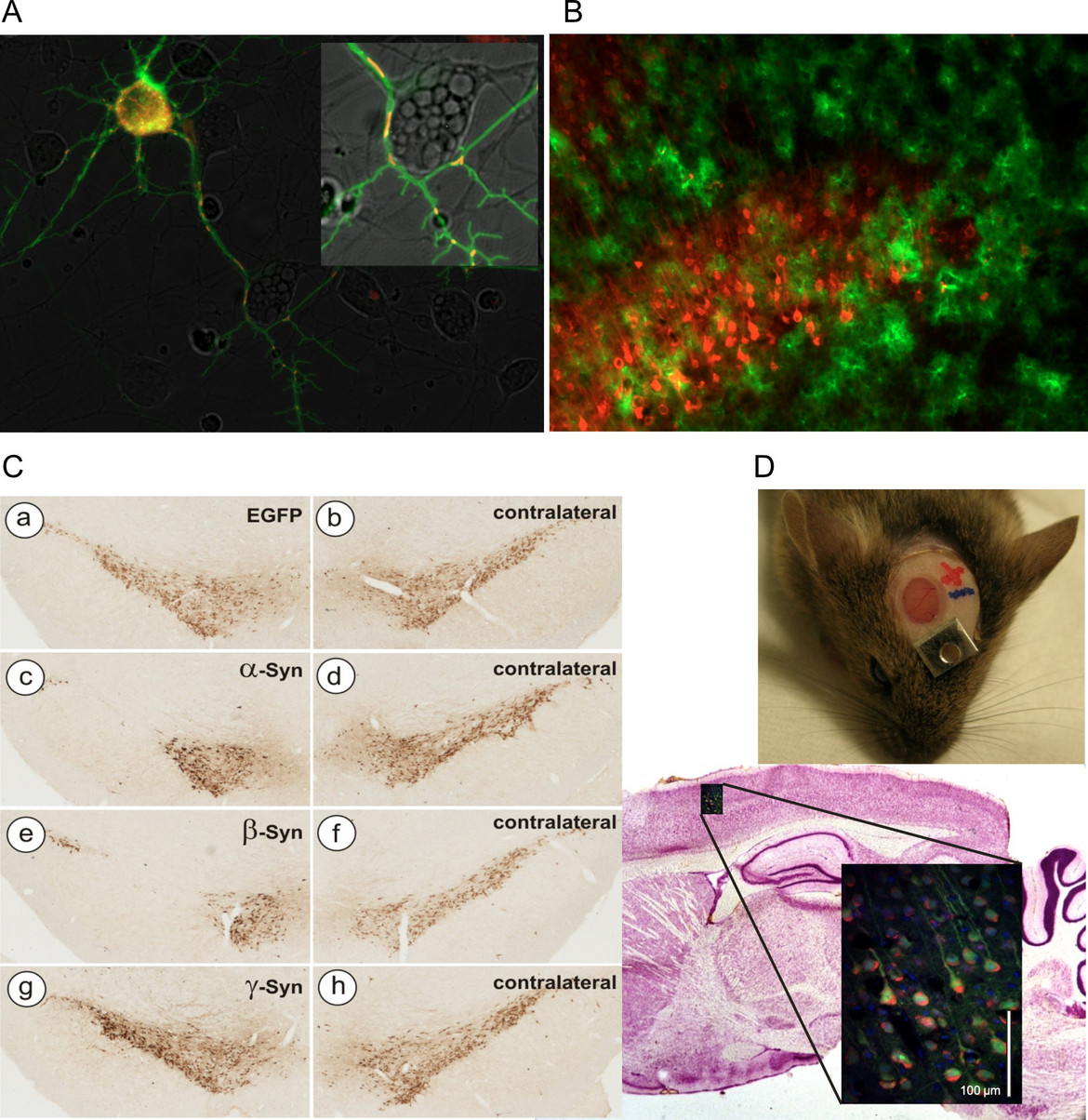
Fig 1: Examples of AAV vector applications routinely used in the lab
A) Concomitant expression of cytoplasmic Ca2+ sensor GCaMP3.5 (green) and mitochondrial Ca2+ sensor RCaMP1e (red) in cultured neurons. Both sensors expressed by AAV-6 vectors under hSyn promoter. B) Mutually exclusive transduction of neurons (AAV-6-hSyn-Cherry, red) and astrocytes (AAV-5-GFAP-EGFP) in mouse cortical layer V. C) Neurodegeneration of nigral dopaminergic neurons after transduction with AAV-2 vectors expressing synuclein proteins (TH immunohistochemistry). D) Imaging of living neurons in mouse brain by 2-photon microscopy through a cranial window.
Pre-clinical gene therapy protocols
Molybdenum Cofactor Deficiency
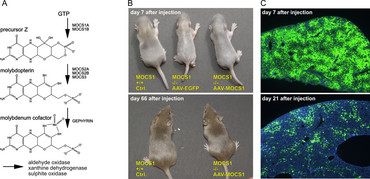
Molybdenum cofactor (MoCo) deficiency is an ultra-rare disease causing early neonatal death. MoCo is synthesized in the liver and functions as an essential co-factor for enzymes detoxifying sulphur components. During pregnancy maternal supply of MoCo prevents lesions, but very soon after birth highly toxic sulphur and xanthine deposits in the brain cause irreversible damages. MOCS1A/1B deficient mice were generated by Jochen Reiss (Dept. of Human Genetics, University Medicine Göttingen), and in a collaborative project we developed an AAV-based gene therapy protocol resulting in healthy animals after liver gene transfer (38). At times of development of this protocol, ultra-rare diseases were out of focus of pharmaceutical industry, and thus no clinical application was initiated. This situation has changed meanwhile (see below), and thus clinical applicability of MoCo gene transfer may still be within reach.
Dual-AAV for large transgenes: treatment of deafness
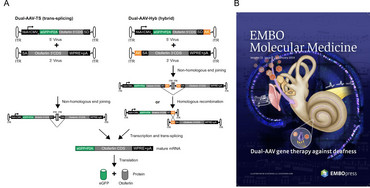
AAV are very small viruses, and thus can package only a quite limited transgene capacity of about 5000 base pairs, of which about 500-1000 bp are necessary for transcriptional control elements like promoter, enhancer and polyadenylation sites. This is unfortunate as several promising gene therapy candidates in sensory systems like retina or inner ear exceed this size by far. Sensory organs are probably the best suited targets for successful gene therapy approaches in the near future, due to their relatively small size, good accessibility and partial protection from immune responses towards the vectors. The Otoferlin protein is defective in a fairly large percentage of people suffering from hereditary hearing loss. Otoferlin is crucially involved into the auditory signal transmission process from inner hair cells to spiral ganglion neurons of the inner ear. As Otoferlin´s cDNA comprises about 6000 bp, it is not suitable to be packaged into a single AAV vector. In addition, we and other groups have demonstrated that vectors with enhanced transgene capacity such as lentiviral or adenoviral vectors are not suitable for inner hair cell transduction.
Thus, in a collaboration with Ellen Reisinger (Dept. of Otorhinolaryngology, University Medicine Göttingen) we exploited a dual AAV vector system in Otoferlin -/- mice, where the 5´-part of Otoferlin was incorporated into one AAV vector, and the 3´-part of Otoferlin was incorporated into a second AAA vector. Appropriately positioned splice sites allowed for reconstitution of full length Otoferlin cDNA in transduced inner hair cells, and enabled a partial but still impressive recovery of auditory function (39). Dual AAV-Otoferlin vectors were out-licensed and are now in highly successful clinical use in the US (with license) and in China (without license J).
Pharmacological control of therapeutic transgene expression
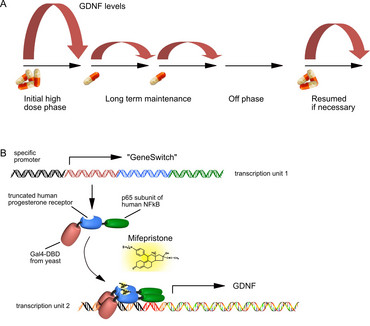
Major neurodegenerative disorders like Alzheimer´s or Parkinson´s disease are characterized by a multifaceted etiology and disease progression. Thus, it is unlikely that a single target will be identified, modification of which might halt disease progression or even revert symptoms in the majority of idiopathic patients. To circumvent this issue, neurotrophic factors (NTFs), that are able to stimulate multiple survival-promoting pathways are considered a valuable option to prevent further loss of neurons and even to restore neuronal functionality. However, the enormous potency of these molecules provokes side effects as well. As NTFs cannot cross the blood-brain-barrier, their expression within the CNS by means of gene therapy is currently considered the optimal route of delivery. As a drawback, gene therapy in its current layout is an irreversible process, meaning that in case of excess supply of NTFs, expression cannot be stopped, nor can the expression level be regulated according to individual patient´s needs.
In order to provide an alternative, we have developed and further optimized an AAV vector system that takes advantage of pharmacological control over transgene expression. The FDA-approved human drug Mifepriston (Mfp), a synthetic steroid, controls expression of the neurotrophic factor GDNF through the so-called “GeneSwitch”, enabling us to tightly control the level and duration of GDNF expression in the brain. The system has proven to work favorably in rat models of Parkinson´s disease, providing robust recovery from motor impairments (40-43).
A regulatory transgene expression system approved for human use will enable a wide portfolio of products, as it allows to address “high risk – high benefit” gene therapy targets, e.g. intracranially expressed single chain antibodies to deplete aggregates in PD and AD, gene editing tools based on Crispr/Cas to directly modify genomic sites, or temporal expression of Yamanaka factors to re-juvenile the aged CNS.
In general, a regulated gene therapy system would enable a highly personalized concept of gene therapy, as it would be no longer an irreversible process.
Safety studies in periphery and CNS of non-human primates are currently underway to prove clinical applicability of this system, so far with promising results.
Synuclein research: a link between blood and brain?
The synuclein protein family consist of three closely related proteins, a-, b-, and g-Synuclein. They all show very little secondary or tertiary structure in solution, and are thus members of the ever-growing family of “unfolded” proteins. a-Synuclein can form protein aggregates under certain conditions, similar to other proteins linked to “aggregopathies” like Tau, Aß or Huntingtin. b-Synuclein was long thought to be an anti-aggregating counterpart to a-Synuclein, but we have recently demonstrated that it forms proteinase K-resistant aggregates in the rodent brain very similar to a-Synuclein (44). a-Synuclein is intimately linked to the etiology of Parkinson´s disease (PD), as mutants and gene multiplications of a-Synuclein are directly causative for parkinsonian symptoms, and aggregations of a-Synuclein are found in all idiopathic PD patients. Throughout the last years we have conducted a series of investigations addressing pathological and physiological functions of the synucleins (2, 6, 7, 18, 22, 45-52). Current research is prompted by the unexpected presence of auto-antibodies against the synucleins in blood of almost every individual. While we have shown that these IgGs cannot serve as reliable biomarkers for Parkinson´s disease (53), recent research has demonstrated that they can induce prominent neurodegeneration when binding to synuclein associated with NMDA receptors on excitatory neurons (54). Given the impaired blood-brain-barrier of many individuals suffering from ageing-associated neurodegenerative diseases, infiltration of blood-borne IgGs into the CNS is likely. Thus, we are about to investigate how relevant this mechanism may be for Parkinsonian patients, by exploiting animal models of IgG transfer into the CNS.
References
- Kashyap R, Balzano M, Lechat B, Lambaerts K, Egea-Jimenez AL, Lembo F, et al. Syntenin-knock out reduces exosome turnover and viral transduction. Sci Rep. 2021;11(1):4083.
- Lodygin D, Hermann M, Schweingruber N, Flügel-Koch C, Watanabe T, Schlosser C, et al. beta-Synuclein-reactive T cells induce autoimmune CNS grey matter degeneration. Nature. 2019;566(7745):503-8.
- Noroozian Z, Xhima K, Huang Y, Kaspar BK, Kügler S, Hynynen K, et al. MRI-Guided Focused Ultrasound for Targeted Delivery of rAAV to the Brain. Methods Mol Biol. 2019;1950:177-97.
- Weber-Adrian D, Kofoed RH, Chan JWY, Silburt J, Noroozian Z, Kügler S, et al. Strategy to enhance transgene expression in proximity of amyloid plaques in a mouse model of Alzheimer's disease. Theranostics. 2019;9(26):8127-37.
- Cuadrado A, Kügler S, Lastres-Becker I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018;14:522-34.
- de Oliveira RM, Vicente Miranda H, Francelle L, Pinho R, Szego EM, Martinho R, et al. The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 2017;15(3):e2000374.
- Lastres-Becker I, Garcia-Yague AJ, Scannevin RH, Casarejos MJ, Kügler S, Rabano A, et al. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson's Disease. Antioxid Redox Signal. 2016;25(2):61-77.
- Wagener RJ, Witte M, Guy J, Mingo-Moreno N, Kügler S, Staiger JF. Thalamocortical Connections Drive Intracortical Activation of Functional Columns in the Mislaminated Reeler Somatosensory Cortex. Cereb Cortex. 2016;26(2):820-37.
- Aveleira CA, Botelho M, Carmo-Silva S, Pascoal JF, Ferreira-Marques M, Nobrega C, et al. Neuropeptide Y stimulates autophagy in hypothalamic neurons. Proc Natl Acad Sci U S A. 2015;112(13):E1642-51.
- Janelidze S, Nordstrom U, Kügler S, Brundin P. Pre-existing immunity to adeno-associated virus (AAV)2 limits transgene expression following intracerebral AAV2-based gene delivery in a 6-hydroxydopamine model of Parkinson's disease. J Gene Med. 2014;16(9-10):300-8.
- Lastres-Becker I, Innamorato NG, Jaworski T, Rabano A, Kügler S, Van Leuven F, et al. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain. 2014;137(Pt 1):78-91.
- Djogo N, Jakovcevski I, Muller C, Lee HJ, Xu JC, Jakovcevski M, et al. Adhesion molecule L1 binds to amyloid beta and reduces Alzheimer's disease pathology in mice. Neurobiol Dis. 2013;56:104-15.
- Akerboom J, Carreras Calderon N, Tian L, Wabnig S, Prigge M, Tolö J, et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci. 2013;6:2.
- Margolis DJ, Lutcke H, Schulz K, Haiss F, Weber B, Kügler S, et al. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat Neurosci. 2012;15(11):1539-46.
- Bahari-Javan S, Maddalena A, Kerimoglu C, Wittnam J, Held T, Bähr M, et al. HDAC1 regulates fear extinction in mice. J Neurosci. 2012;32(15):5062-73.
- Minderer M, Liu W, Sumanovski LT, Kügler S, Helmchen F, Margolis DJ. Chronic imaging of cortical sensory map dynamics using a genetically encoded calcium indicator. J Physiol. 2012;590(Pt 1):99-107.
- Jaworski T, Dewachter I, Lechat B, Gees M, Kremer A, Demedts D, et al. GSK-3alpha/beta kinases and amyloid production in vivo. Nature. 2011;480(7376):E4-5; discussion E6.
- Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol. 2011;194(1):49-60.
- Schaechinger TJ, Gorbunov D, Halaszovich CR, Moser T, Kügler S, Fakler B, et al. A synthetic prestin reveals protein domains and molecular operation of outer hair cell piezoelectricity. EMBO J. 2011;30(14):2793-804.
- Sousa-Ferreira L, Alvaro AR, Aveleira C, Santana M, Brandao I, Kügler S, et al. Proliferative hypothalamic neurospheres express NPY, AGRP, POMC, CART and Orexin-A and differentiate to functional neurons. PLoS One. 2011;6(5):e19745.
- Mironov SL, Skorova EY, Kügler S. Epac-mediated cAMP-signalling in the mouse model of Rett Syndrome. Neuropharmacology. 2011;60(6):869-77.
- Garrido M, Tereshchenko Y, Zhevtsova Z, Taschenberger G, Bähr M, Kügler S. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathol. 2011;121(4):475-85.
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, et al. Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci. 2010;30(46):15654-63.
- Jaworski T, Kügler S, Van Leuven F. Modeling of tau-mediated synaptic and neuronal degeneration in Alzheimer's disease. Int J Alzheimers Dis. 2010;2010.
- Chen J, Joon Lee H, Jakovcevski I, Shah R, Bhagat N, Loers G, et al. The extracellular matrix glycoprotein tenascin-C is beneficial for spinal cord regeneration. Mol Ther. 2010;18(10):1769-77.
- Lütcke H, Murayama M, Hahn T, Margolis DJ, Astori S, Zum Alten Borgloh SM, et al. Optical recording of neuronal activity with a genetically-encoded calcium indicator in anesthetized and freely moving mice. Front Neural Circuits. 2010;4:9.
- Jaworski T, Dewachter I, Seymour CM, Borghgraef P, Devijver H, Kügler S, et al. Alzheimer's disease: old problem, new views from transgenic and viral models. Biochim Biophys Acta. 2010;1802(10):808-18.
- Jaworski T, Dewachter I, Lechat B, Croes S, Termont A, Demedts D, et al. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS One. 2009;4(10):e7280.
- Mironov SL, Skorova E, Hartelt N, Mironova LA, Hasan MT, Kügler S. Remodelling of the respiratory network in a mouse model of Rett syndrome depends on brain-derived neurotrophic factor regulated slow calcium buffering. J Physiol. 2009;587(Pt 11):2473-85.
- Wallace DJ, Meyer zum Alten Borgloh S, Astori S, Yang Y, Bausen M, Kügler S, et al. Single-spike detection in vitro and in vivo with a genetic Ca2+ sensor. Nat Methods. 2008;5(9):797-804.
- Hartelt N, Skorova E, Manzke T, Suhr M, Mironova L, Kügler S, et al. Imaging of respiratory network topology in living brainstem slices. Mol Cell Neurosci. 2008;37(3):425-31.
- Tomita H, Sugano E, Yawo H, Ishizuka T, Isago H, Narikawa S, et al. Restoration of visual response in aged dystrophic RCS rats using AAV-mediated channelopsin-2 gene transfer. Invest Ophthalmol Vis Sci. 2007;48(8):3821-6.
- Shevtsova Z, Malik I, Garrido M, Schöll U, Bähr M, Kügler S. Potentiation of in vivo neuroprotection by BclX(L) and GDNF co-expression depends on post-lesion time in deafferentiated CNS neurons. Gene Ther. 2006;13(22):1569-78.
- Malik JM, Shevtsova Z, Bähr M, Kügler S. Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL gene transfer. Mol Ther. 2005;11(3):373-81.
- Reisinger E, Bresee C, Neef J, Nair R, Reuter K, Bulankina A, et al. Probing the functional equivalence of otoferlin and synaptotagmin 1 in exocytosis. J Neurosci. 2011;31(13):4886-95.
- Knauer C, Haltern H, Schoger E, Kügler S, Roos L, Zelarayan LC, et al. Preclinical evaluation of CRISPR-based therapies for Noonan syndrome caused by deep-intronic LZTR1 variants. Mol Ther Nucleic Acids. 2024;35(1):102123.
- Drummer C, Vogt EJ, Heistermann M, Roshani B, Becker T, Matz-Rensing K, et al. Generation and Breeding of EGFP-Transgenic Marmoset Monkeys: Cell Chimerism and Implications for Disease Modeling. Cells. 2021;10(3).
- Kügler S, Hahnewald R, Garrido M, Reiss J. Long-term rescue of a lethal inherited disease by adeno-associated virus-mediated gene transfer in a mouse model of molybdenum-cofactor deficiency. Am J Hum Genet. 2007;80(2):291-7.
- Al-Moyed H, Cepeda AP, Jung S, Moser T, Kügler S, Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. 2019;11(1).
- Maddalena A, Tereshchenko J, Bähr M, Kügler S. Adeno-associated Virus-mediated, Mifepristone-regulated Transgene Expression in the Brain. Mol Ther Nucleic Acids. 2013;2:e106.
- Cheng S, van Gaalen MM, Bähr M, Garea-Rodriguez E, Kügler S. Optimized pharmacological control over the AAV-Gene-Switch vector for regulable gene therapy. Mol Ther Methods Clin Dev. 2021;23:1-10.
- Cheng S, Tereshchenko J, Zimmer V, Vachey G, Pythoud C, Rey M, et al. Therapeutic efficacy of regulable GDNF expression for Huntington's and Parkinson's disease by a high-induction, background-free "GeneSwitch" vector. Exp Neurol. 2018;309:79-90.
- Tereshchenko J, Maddalena A, Bähr M, Kügler S. Pharmacologically controlled, discontinuous GDNF gene therapy restores motor function in a rat model of Parkinson's disease. Neurobiol Dis. 2014;65:35-42.
- Taschenberger G, Tolö J, Tereshchenko J, Akerboom J, Wales P, Benz R, et al. beta-synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann Neurol. 2013;74(1):109-18.
- Leite K, Garg P, Spitzner FP, Guerin Darvas S, Bähr M, Priesemann V, et al. alpha-Synuclein Impacts on Intrinsic Neuronal Network Activity Through Reduced Levels of Cyclic AMP and Diminished Numbers of Active Presynaptic Terminals. Front Mol Neurosci. 2022;15:868790.
- Psol M, Darvas SG, Leite K, Mahajani SU, Bähr M, Kügler S. Dementia with Lewy bodies-associated ss-synuclein mutations V70M and P123H cause mutation-specific neuropathological lesions. Hum Mol Genet. 2021;30(3-4):247-64.
- Raina A, Leite K, Guerin S, Mahajani SU, Chakrabarti KS, Voll D, et al. Dopamine promotes the neurodegenerative potential of beta-synuclein. J Neurochem. 2021;156(5):674-91.
- Tolö J, Taschenberger G, Leite K, Stahlberg MA, Spehlbrink G, Kues J, et al. Pathophysiological Consequences of Neuronal alpha-Synuclein Overexpression: Impacts on Ion Homeostasis, Stress Signaling, Mitochondrial Integrity, and Electrical Activity. Front Mol Neurosci. 2018;11:49.
- Eckermann K, Kügler S, Bähr M. Dimerization propensities of Synucleins are not predictive for Synuclein aggregation. Biochim Biophys Acta. 2015;1852(8):1658-64.
- Kunadt M, Eckermann K, Stuendl A, Gong J, Russo B, Strauss K, et al. Extracellular vesicle sorting of alpha-Synuclein is regulated by sumoylation. Acta Neuropathol. 2015;129(5):695-713.
- Tönges L, Szego EM, Hause P, Saal KA, Tatenhorst L, Koch JC, et al. Alpha-synuclein mutations impair axonal regeneration in models of Parkinson's disease. Front Aging Neurosci. 2014;6:239.
- Karpinar DP, Balija MB, Kügler S, Opazo F, Rezaei-Ghaleh N, Wender N, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 2009;28(20):3256-68.
- Garg P, Maass F, Sundaram SM, Mollenhauer B, Mahajani S, van Riesen C, et al. The relevance of synuclein autoantibodies as a biomarker for Parkinson's disease. Mol Cell Neurosci. 2022;121:103746.
- Garg P, Würtz F, Hobbie F, Buttgereit K, Aich A, Leite K, et al. Human serum-derived alpha-synuclein auto-antibodies mediate NMDA receptor-dependent degeneration of CNS neurons. J Neuroinflammation. 2024;21(1):62.
Contact
contact information
- e-mail address: sebastian.kuegler(at)med.uni-goettingen.de